OVERVIEW
Our research addresses biological and medical challenges from single molecules to the genome with high performance computing and theory. Specific areas of research include protein-ligand interactions for drug discovery, RNA structure prediction and biomaterials design. In collaboration with other experimental groups, we utilize computer modeling and simulations to understand these complex biomolecular systems and to discover molecules for treating disease and improving human health.
In the areas of software development, we are contributors to molecular modeling software TINKER and AMBER. Our physical models (polarizable force fields) are also available through FFX and OpenMM.
COMPUTATIONAL METHODS AND SOFTWARE DEVELOPMENT
One of our main research directions is to develop cutting edge physical models (polarizable AMOEBA force field) and software for molecular modeling and design. We develop force fields for proteins, nucleic acids and other biomolecules that enable us to accurately predict biomolecular structures, dynamics and interactions in silico. In collaboration with with Prof. Jay Ponder (Wash Univ in St. Louis), Jean-Philips Piquemal (Sorbonne Univ), Andres Cisneros (UNT), Mike Schnieders (Univ. Iowa) and Wei Yang (Florida State Univ), we develop the molecular modeling platform including Tinker, Tinker-HP, Tinker-GPU. This research is supported by National Institute of Health.
https://github.com/TinkerTools
DRUG DISCOVERY
By prediction interactions of biological targets (e.g. proteins and nucleic acids) with specific ligand molecules, computer simulations can be applied to accelerate drug discovery. We are part of The Targeted Therapeutic Drug Discovery & Development Program (TTDDDP) , supported by Cancer Prevention Research Institute of Texas.
PROTEIN-LIGAND RECOGNITION
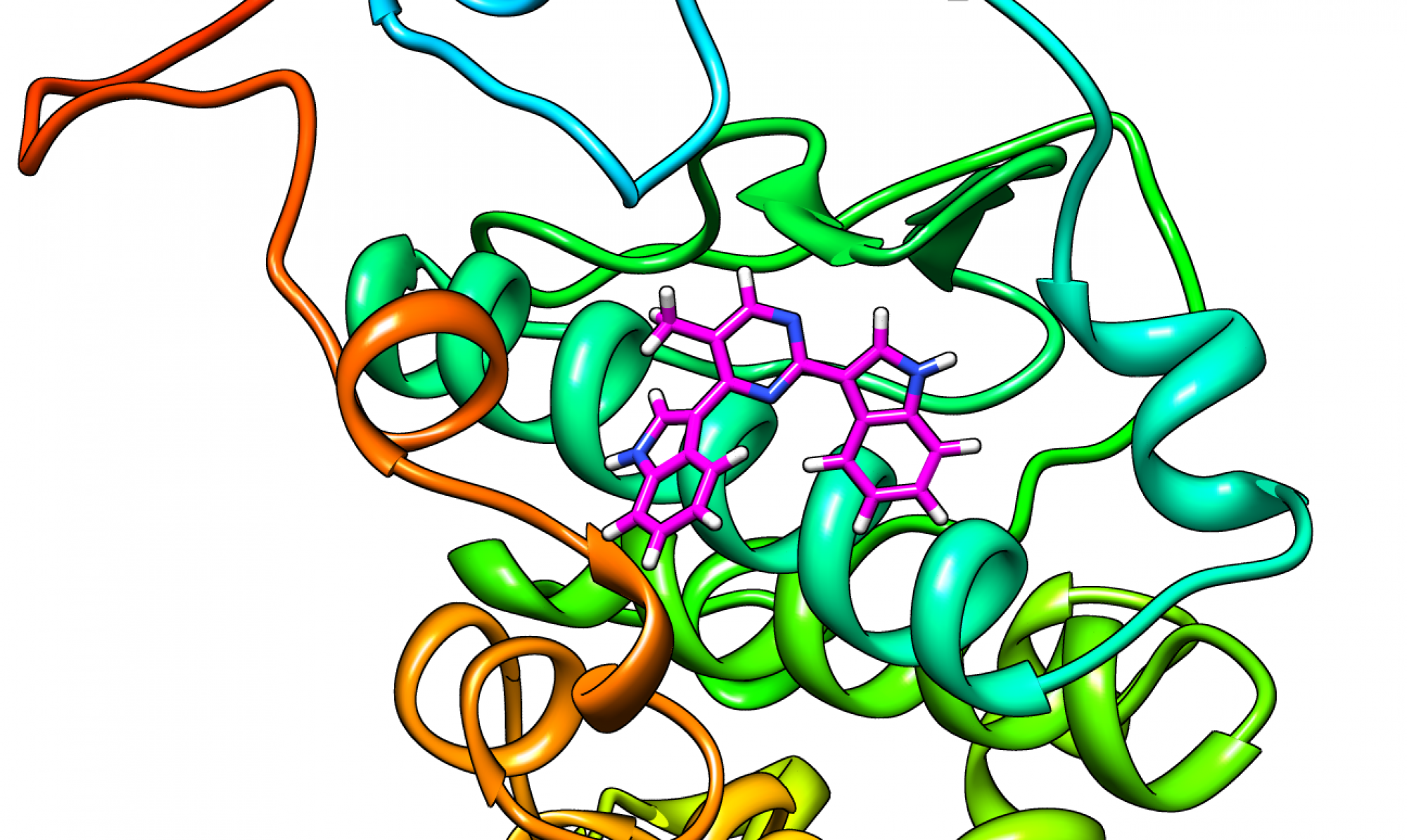
We are interested in understanding the molecular driving forces for protein-ligand recognition using modeling and simulations, and developing novel molecular probes and therapeutics.
Working with Prof. Kevin Dalby in Med Chem, UT School of Pharmacy, we are examining proteins in the MAPK signaling pathway, which are involved in cell proliferation, apoptosis and resistance to cancer therapy. Our work helps understand how these proteins work and develop novel inhibitors and drug candidates.
A movie on cell MAPK signaling.
MULTISCALE MODELING of RNA
Nucleic acids are essential molecules in cell. Their function depends on their structures and interactions with other molecules in cell. In addition to developing atomic model (AMOEBA) for nucleic acid modeling, we are also interested in coarse-grained model (RACER) for efficient ab initio prediction of RNA folding. This research has been supported by Welch Foundation.
Folding of U2/U6 snRNA using a coarse-grained model developed in the lab.
ORGANIC CRYSTAL STRUCTURE & PROPERTY PREDICTION
Simulation of Aspirin crystal structure using advanced sampling (Orthogonal Space Sampling), in collaboration with Prof Wei Yang (FSU).
SELF-ASSEMBLY MATERIALS
In collaboration with Prof. Laura Suggs of UT BME, we investigated the self-assembly mechanism of Fmoc-AA fibril to assist the development of new mimetics for tissue engineering applications.